碳氢键是有机分子中最基本、最普遍的化学键。由于复杂的有机分子中通常含有多个不同类型的碳氢键,如何精准且高效地活化某一个特定位置是该领域的难点。在配体骨架上引入合适的导向基团被认为是一种有效的策略,通过催化剂与底物之间的非共价相互作用,例如氢键相互作用、离子对相互作用、静电相互作用和路易斯酸碱对相互作用,使过渡金属催化剂在碳氢活化反应中具有良好的选择性。尽管使用导向基团策略取得了许多成就,但催化反应的活性通常不够高,无法实际使用,需要进一步研究以提高其催化活性。最近,计算化学研究所雷鸣课题组与河北大学李龙飞课题组合作,通过DFT理论计算提出了一种结合导向基团、配位不饱和金属中心和阳离子特性多种策略的阳离子催化剂,用于提高碳氢硼化反应中的活性和选择性,并揭示了邻位碳氢硼化区域选择性的起源,相关成果以全文发表J. Org. Chem.上 (https://doi.org/10.1021/acs.joc.1c02070)。
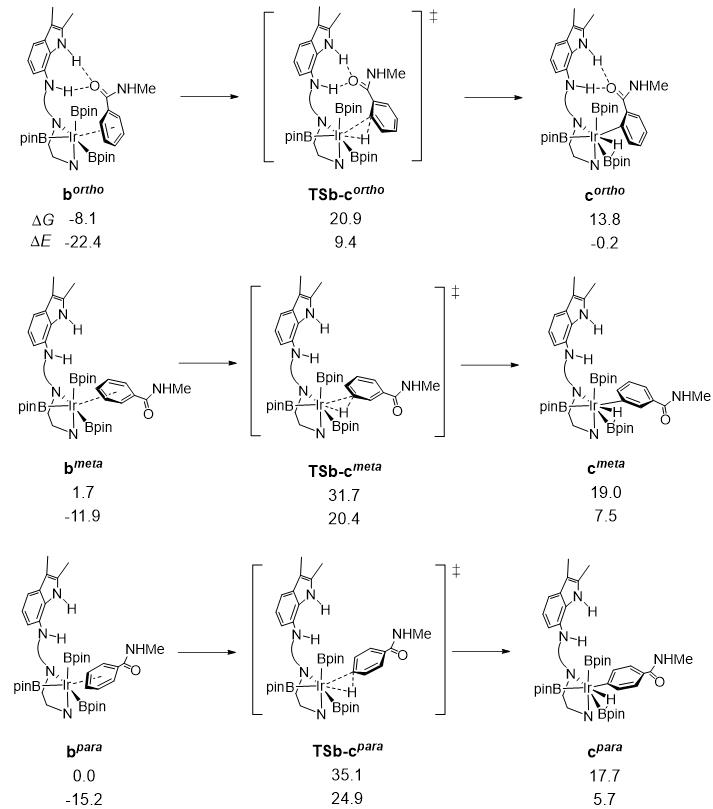
图1. 原中性Ir催化剂催化碳氢键裂解反应步骤的吉布斯自由能和电子能(单位:kcal/mol)。
首先,我们计算研究了中性Ir催化剂催化碳氢硼化反应过程。如图1所示,在氢键裂解反应步骤中,碳氢键键裂解过渡态TSb-cortho, TSb-cmeta和TSb-cpara的相对能量分别为20.9、31.7和35.1 kcal/mol,表明邻位硼化途径是最有利的。TSb-cortho的稳定性来源于配体上吲哚酰胺和底物的羰基氧原子之间的氢键相互作用,导致邻位选择性较为优势。然而,有利的邻位碳氢硼化反应路径的反应能垒为29.0 kcal/mol,实验上也报道该反应的时间较长。
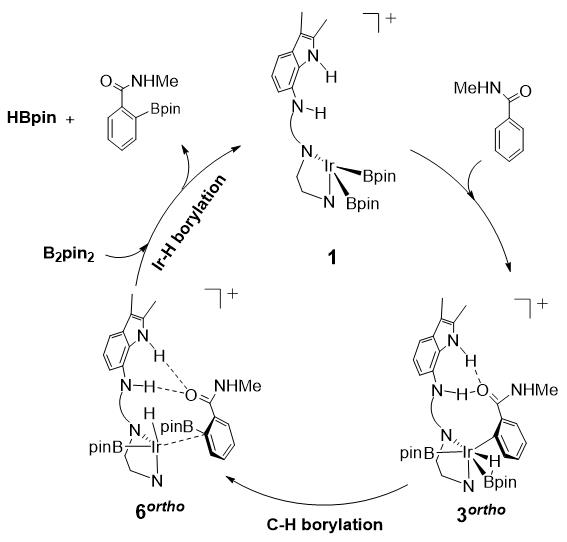
图2. 阳离子催化剂1催化N-甲基苯甲酰胺邻位碳氢硼化的机理。
如图2所示,我们设计了一种具有阳离子Ir中心和两个Bpin配体的新型催化剂1,其催化N-甲基苯甲酰胺邻位碳氢硼化路径包括C-H硼化(1→3ortho→6ortho)和Ir-H硼化(6ortho→1)反应步骤。
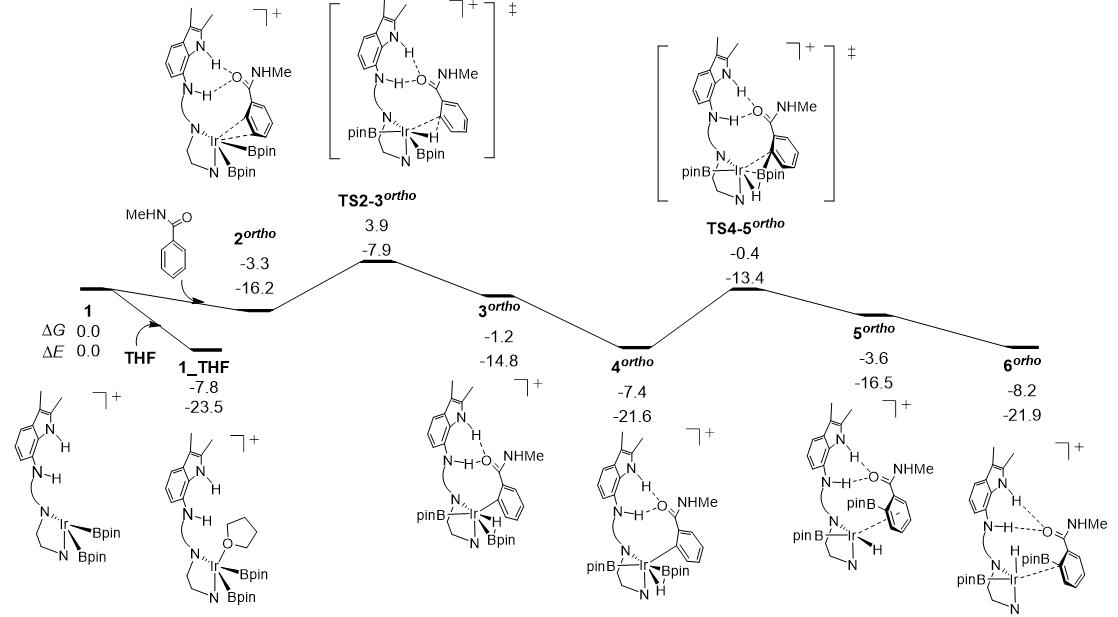
图3.阳离子催化剂1对 N-甲基苯甲酰胺邻位C-H硼化过程的反应自由能面图(反应路径中各驻点下数字分别为相对催化组分1的相对自由能和电子能量,能量单位:kcal/mol)。
图3给出了化剂1催化的碳氢硼化过程的反应自由能面图,其中,包含了碳氢键断裂(1→3ortho)和碳硼键形成(3ortho→6ortho)步骤。计算结果表明,反应底物苯环上邻间对位上碳氢键裂解过渡态TS2-3ortho,TS2-3meta和TS2-3para)的相对自由能分别为3.9,6.2和8.0 kcal/mol。因此,邻位途径是最有利的,具有较高的选择性。
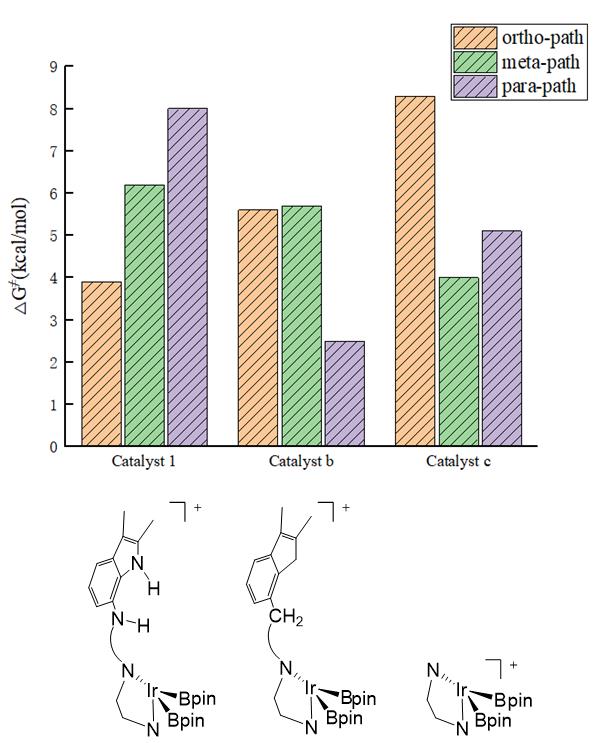
图4.设计和改性Ir配合物催化不同区位C-H键活化反应路径中过渡态相对吉布斯自由能示意图(单位:kcal/mol)。
为了证明导向基团的作用,我们进一步进行了催化剂1的改性设计,并研究了区域选择性的变化(见图4)。计算结果表明,通过使用亚甲基取代氨基以及脱去配体上的吲哚酰胺基团,邻位选择性都变得不再优势。配体上的吲哚酰胺与底物羰基之间的氢键相互作用导致底物的邻位碳氢键靠近铱中心,从而有助于邻位选择性。
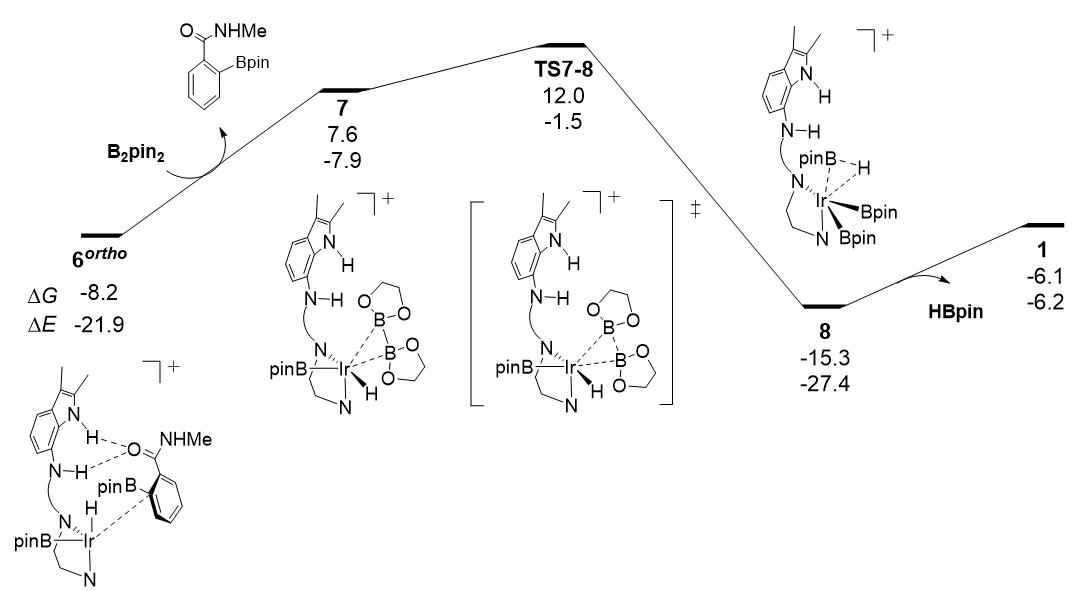
图5.Ir-H硼化过程中的反应自由能面图(反应路径中各驻点下数字分别为相对催化组分1的相对自由能和电子能量,能量单位:kcal/mol)。
随后,本文对Ir-H硼化过程进行了研究(图5)。有意思的是,Ir-H与B2pin2硼化过程的能量跨度高达20.2 kcal/mol,而原中性催化剂与B2pin2的Ir-H硼化过程较容易,反应能垒为7.2 kcal/mol。如图6所示,与中性催化剂相比,阳离子催化剂在催化C-H键裂解中具有较高的活性,但在B-B键裂解过程中中性催化剂具有更高的活性。
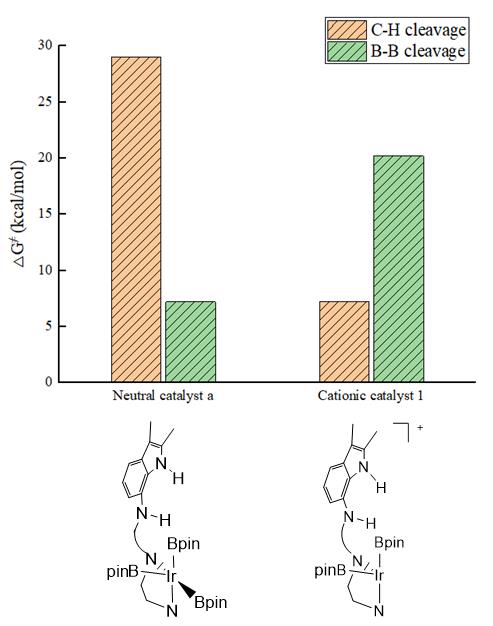
图6.中性和阳离子Ir配合物催化C-H和B-B键断裂的吉布斯自由能示意图(单位:kcal/mol)。
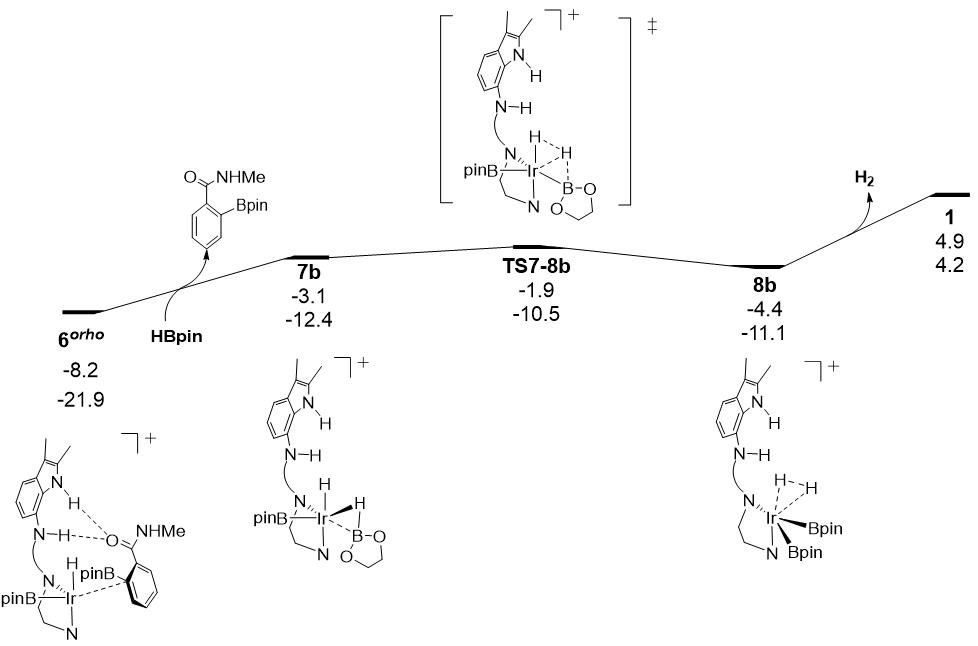
图7.使用HBpin设计的Ir-H硼化过程的反应自由能面图(反应路径中各驻点下数字分别为相对催化组分1的相对自由能和电子能量,能量单位:kcal/mol)。
为了解决阳离子铱催化剂在Ir-H硼化中活性低的问题,我们转向使用HBpin做为硼化来源探索该反应途径(见图7),计算结果表明使用HBpin的Ir-H硼化的反应能垒仅为6.3 kcal/mol,远低于使用B2pin2 (20.2 kcal/mol)的Ir-H硼化反应能垒。
综上所述,本文设计了一种在碳氢硼化反应中保持选择性并提高反应活性的阳离子Ir催化剂,并对其反应机理进行了探究,指出碳氢裂解步骤为整个催化反应循环中的区域选择性决定步骤,邻位碳氢硼化途径比间位和对位更有利,邻位C-H键活化反应能垒为7.2 kcal/mol,表明该过程中阳离子Ir催化剂比中性Ir催化剂具有更高的催化活性。此外,本文进一步进行催化剂的改性与设计,证明了该反应的邻位选择性归因于导向基团和底物之间的氢键相互作用(尽管邻位在空间和电子上是不利的)。本文的研究为面向碳氢硼化反应的具有高活性高选择性的过渡金属催化剂的分子设计提供了一定的理论参考。该文第一作者为计算化学所2019级硕士研究生刘翀同学,此工作获得了国家自然科学基金面上基金项目的支持。