将二氧化碳转化为燃料、聚合物和化学品是合成高附加值产品和储能材料的一种极具吸引力的策略。在众多储氢产品中,甲醇及其衍生物因其高储氢能力而备受科学家关注。然而,二氧化碳转化为甲醇及其衍生物需要苛刻的反应条件,目前还无法解决。因此,开发高效的二氧化碳转化催化剂,特别是廉价金属催化剂具有重要的意义。近期,计算化学研究所雷鸣教授与厦门大学曹泽星教授课题组合作,采用密度泛函理论(DFT)计算研究了锰钳型配合物([Mn(Ph2PCH2SiMe2)2N(CO)2])催化二氧化碳硼氢化的反应机理,并揭示了反位配体对该Mn-PNP配合物的催化活性具有显著地调控作用,相关成果以全文发表在Inorg. Chem.上(https://pubs.acs.org/doi/10.1021/acs.inorgchem.2c00285)。
如图1所示,锰配合物A1作为催化活性物种,其催化二氧化碳硼氢化反应主要包括羰基缔合路径和羰基解离路径。

图1. Mn-PNP催化二氧化碳硼氢化的反应机理示意图。
首先,对羰基缔合路径的反应机理进行探究,主要包括三个催化循环:二氧化碳硼氢化、HCOOBpin硼氢化以及HCHO硼氢化。计算结果表明,中间产物HCOOBpin的硼氢化过程(A3→TSA6)是整个羰基缔合路径的决速步骤,自由能垒为27.0 kcal/mol (图2)。另外,Leitner等人的实验结果表明溶剂四氢呋喃和甲苯会使反应的产率和转化数显著降低,我们对溶剂效应的探究表明该反应在有机溶剂四氢呋喃和甲苯中的自由能分布比气相更高,决速步的自由能垒分别为33.7和31.9 kcal/mol。这与Leitner等人的实验结果是一致的。
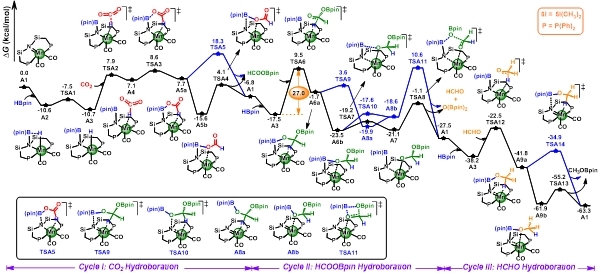
图2. Mn-PNP催化二氧化碳硼氢化羰基缔合路径的自由能面图(单位:kcal/mol)。
另外,羰基缔合路径中中间体A3的高稳定性导致氢转移的能垒较高,因此,作者提出了羰基解离路径以绕过物种A3的产生。计算结果表明,羰基释放是羰基解离路径的决速步骤,自由能垒为35.2 kcal/mol,比羰基缔合路径决速步能垒高8.2 kcal/mol。同时,探究了溶剂效应对羰基解离路径的影响,计算结果与实验结果一致。

图3. Mn-PNP催化二氧化碳硼氢化羰基解离路径的自由能面图(单位:kcal/mol)。
之前的研究表明,反式配体在加氢反应中起着重要作用,这可能会影响M-H键的键长以及金属上氢负离子的电荷。在这项工作中,探究了具有不同X配体(X = CO、Br、H、PH3)的Mn-PNP 配合物的反位配体效应对二氧化碳硼氢化的催化活性(图4)。当CO 配体被H或Br等阴离子配体取代,Mn-PNP 配合物可能有两种可能的电子结构:一是金属中心的氧化态保持不变,配合物带负电;另一种是配合物呈中性,金属中心的氧化态从Mn(I)增加到Mn(II)。计算结果表明,反位配体可以改变反应的速率决定步骤,Mn-PNP配合物[Mn(I)-H]-和[Mn(II)-H]比[Mn(I)-CO]具有更高的催化活性,表明H配体对配合物的催化活性有很好的调控作用,其中,Mn(I)-阴离子配合物具有更好的催化活性。因此,以H为反位配体的[Mn(I)-H]-配合物是一种在温和条件下催化二氧化碳硼氢化的高效催化剂。
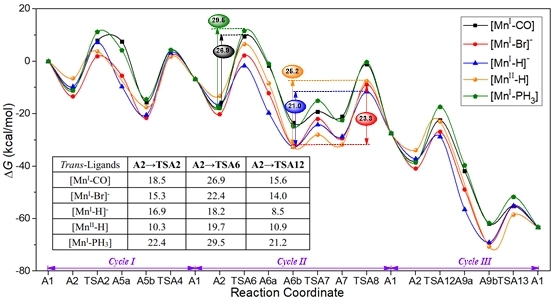
图4. 具有不同反位配体的Mn-PNP配合物催化二氧化碳硼氢化的自由能面图 (单位:kcal/mol)。
综上所述,采用DFT 方法研究了锰钳型配合物催化二氧化碳硼氢化的反应机理。分别研究了羰基缔合和羰基解离机理,每种机理包括三个催化循环:二氧化碳硼氢化、HCOOBpin硼氢化以及HCHO硼氢化。计算结果表明,Mn-PNP催化二氧化碳硼氢化羰基缔合机理的能量跨度为27.0 kcal/mol,而羰基解离机理的速率决定步骤是CO配体释放,能垒为35.2 kcal/mol。因此,羰基缔合机理更有利。此外,本文还探讨了不同反位配体对锰配合物催化活性的影响,计算结果表明,[Mn(I)-H]-配合物的催化活性最高,能量跨度为21.0 kcal/mol。本工作将为廉价金属催化剂的设计和后期羰基硼氢化机理的研究提供一定的理论参考。该工作的第一作者为计算化学所2021届硕士毕业生张林同学(现为厦门大学化学化工学院2021级博士研究生),此工作获得了国家自然科学基金面上基金项目的支持。