电化学水分解制氢是解决能源危机和环境污染的重要技术之一,可以通过将水分解为氢气和氧气,从而实现绿色氢能的可持续生产。在这一过程中,阳极的析氧反应(OER)作为关键步骤,因其复杂的四电子转移过程和较高的过电位,成为制约整体效率的主要瓶颈。目前,贵金属催化剂(如RuO2、IrO2)虽表现出优异的OER活性,但其高昂的成本和稀缺性限制了大规模应用。层状双氢氧化物(LDH)凭借可调的电子结构和低成本优势,被视为理想替代材料,通过金属掺杂策略优化LDH的电催化OER反应性能是当前的研究热点之一。北京化工大学计算化学研究所雷鸣课题组采用密度泛函理论(DFT)方法计算研究了11种过渡金属 (TM = V、Cr、Mn、Ru、Rh、Pd、Ag、Os、Ir、Pt、Au) 掺杂的四种LDH模型(见如图1的TMII/TMIII@M3N-LDH(110),M3N-LDH(110)=Co3Fe-LDH(110), Ni3Fe-LDH(110), Ni3Co-LDH(110)和Ni3Mn-LDH(110))的电催化OER反应性能,结合电子结构分析,构建了ΔG*O-ΔG*OH和-ηOER的火山图关系,从88个构建的TMII/TMIII@M3N-LDH(110)催化体系中筛选出7种具有潜在高催化活性的OER电催化剂,其中铂(PtIII)和钌(RuII)掺杂的钴铁层状双氢氧化物(Co3Fe-LDH(110))理论计算的过电位值(ηOER)低至0.20 V,较商用RuO2催化剂的理论计算ηOER(0.37 V)减小了约46%。该研究为设计与开发低成本、高性能过渡金属掺杂二维材料的OER电催化剂提供了重要的理论支撑。相关成果以全文发表在Appl. Surf. Sci.上 (https://doi.org/10.1016/j.apsusc.2024.161233)。
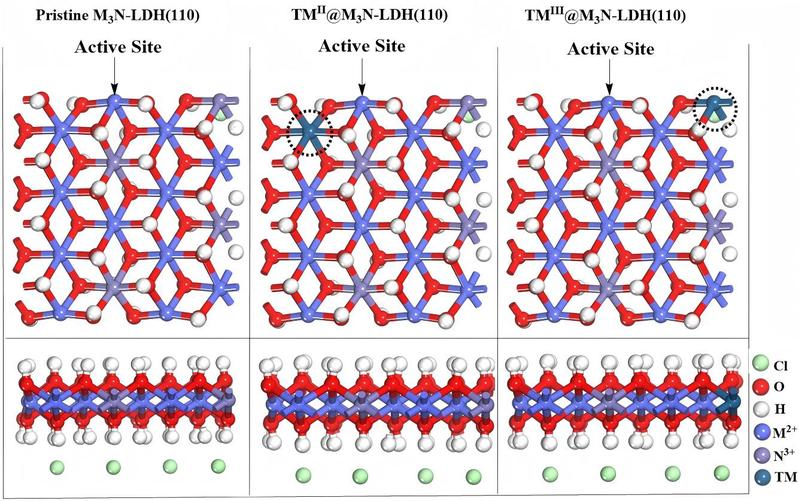
图1. 原始M3N-LDH(110),过渡金属TM掺杂LDH(TMII@M3N-LDH(110)和TMIII@M3N-LDH(110))的计算优化结构示意图(圆圈表示TM在LDH中的掺杂位点)。
掺杂原子的团聚会显著降低掺杂体系的催化活性。为评估掺杂原子的分散稳定性,我们采用结合能(Eb)和内聚能(Ec)判据。Eb反映单个掺杂原子与基底的结合强度(Eb越负,表明结合越稳定)。Ec表征块体金属中原子间的内聚力。当满足Eb < Ec时(即Eb比Ec更负),说明掺杂原子与基底结合强度超过其自身金属的内聚力。此时,掺杂原子倾向于嵌入基底表面,而非团聚为金属簇。本研究系统计算了88种过渡金属掺杂LDH体系的Eb与Ec(见图2),计算结果表明,大部分TM掺杂体系满足Eb < Ec的稳定性要求,可实现TM在基底上的原子级分散。值得注意的是,所有掺杂体系的Eb均为负值(Eb < 0.0 eV),表明TM原子可稳定锚定在LDH基底上。
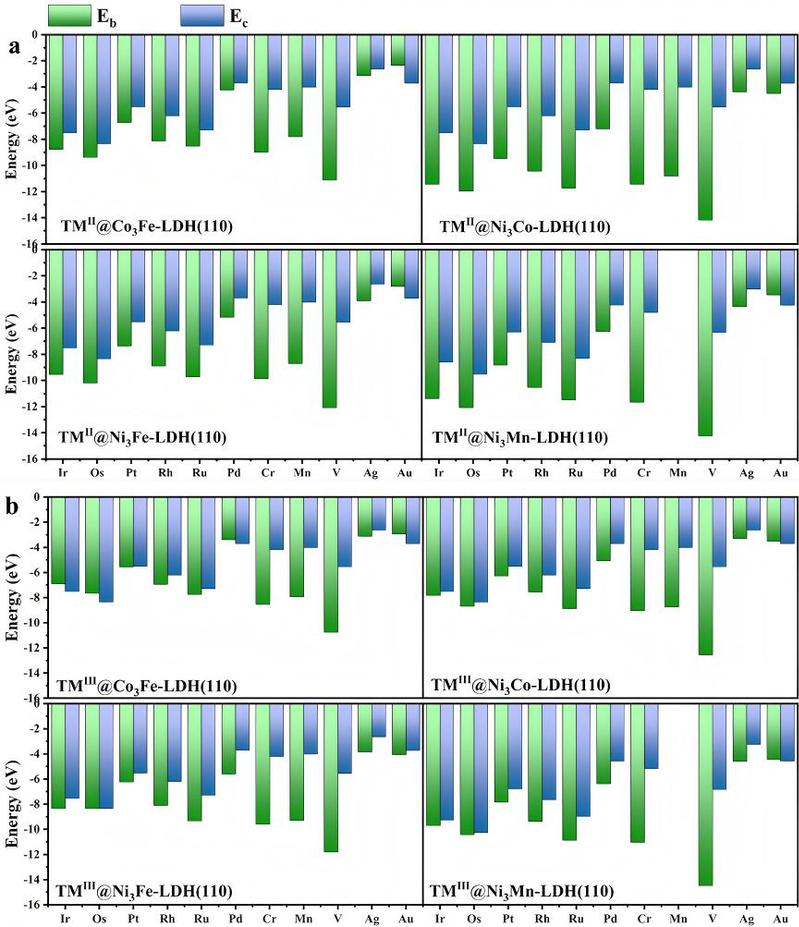
图2. TM掺杂LDH体系(a) TMII@M3N-LDH(110), (b) TMIII@M3N-LDH(110)的结合能(Eb)与块体内聚能(Ec)对比(单位:eV)。
随后,本研究计算了88个构建的过渡金属TM掺杂M3N-LDH(110)体系电催化OER反应的全路径,并构建了ΔG*O-ΔG*OH与-ηOER之间的OER反应火山图(见图3a-3e)。可以看出,对此类TM掺杂M3N-LDH(110)催化体系,当*O与*OH在表面的吸附自由能差值ΔG*O-ΔG*OH趋近于1.38 eV时(该值对应最优中间体吸附强度),OER电催化活性最好。以原始Co3Fe-LDH(110)为基准(ηOER=0.33 V),本研究从88个TM掺杂LDH体系中筛选出7种具有潜在高活性的OER电催化剂,分别为:AgⅢ@Co3Fe-LDH(110) (ηOER=0.31 V) < MnII@Co3Fe-LDH(110) (ηOER=0.28 V) < PdII@Co3Fe-LDH(110) (ηOER=0.27 V), RhII@Co3Fe-LDH(110) (ηOER=0.27 V) < CrⅢ@Co3Fe-LDH(110) (ηOER=0.23 V) < PtⅢ@Co3Fe-LDH(110), RuII@Co3Fe-LDH(110) (ηOER=0.20 V),其中PtIII@Co3Fe-LDH(110)与RuII@Co3Fe-LDH(110)电催化剂位于火山顶点附近,具有最优的OER电催化活性。

图3. (a) TMII/TMIII@Co3Fe-LDH(110),(b) TMII/TMIII@Ni3Co-LDH(110),(c) TMII/TMIII@Ni3Fe-LDH(110),(d) TMII/TMIII@Ni3Mn-LDH(110)和(e) 上述四类TMII/TMIII@M3N-LDH(110)体系催化OER反应的理论计算过电位(-ηOER)与步骤3和步骤2之间的吉布斯自由能差(ΔG*O-ΔG*OH)之间的火山图关系。
为了揭示TM掺杂M3N-LDH(110)体系的电子调控机制,本研究以Co3Fe-LDH(110)为基准体系,选择d带中心(εd)作为反应描述符,系统分析其与过电位(-ηOER)与εd之间的关系(见图4)。对于TMII@Co3Fe-LDH(110)体系,εd与-ηOER在一定范围内存在线性关系,当εd接近费米能级(EF),ηOER减小。对于TMIII@Co3Fe-LDH(110)催化剂,-ηOER与εd呈现火山图的关系,当εd值约为1.35 eV时,其OER电催化性能最好。同时,我们对筛选出的PtIII@Co3Fe-LDH(110)与RuII@Co3Fe-LDH(110)进行了态密度(DOS)分析,分析结果指出,经过TM掺杂后,这两种体系的带隙相较于原始Co3Fe-LDH(110)都显著降低,且费米能级附近的电子态主要由掺杂离子的峰构成,这表明通过TM掺杂M3N-LDH(110)体系可以提高其导电性,从而提升了TM掺杂LDH体系的OER电催化性能。
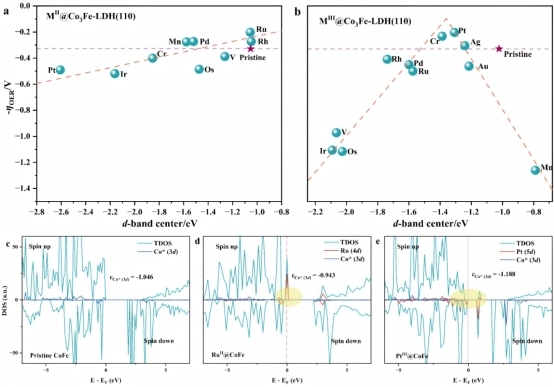
图4. (a) TMII@Co3Fe-LDH(110)和(b) TMIII@Co3Fe-LDH(110)体系d带中心(εd)与过电位(-ηOER)之间的关系图,(c) 原始Co3Fe-LDH(110),(d) RuII@Co3Fe-LDH(110)和(e) PtIII@Co3Fe-LDH(110)催化体系的态密度(DOS)图(虚线表示费米能级)。
综上所述,本研究采用DFT方法,理论设计并系统研究了过渡金属TM掺杂M3N-LDH(110)体系电催化OER反应活性,揭示了TM掺杂LDH体系的电子调控机制,构建了催化体系的ΔG*O-ΔG*OH与-ηOER之间的火山图关系,并从设计的88个TMII/TMIII@M3N-LDH(110)催化体系中筛选出了7个稳定且具有潜在高活性的OER电催化剂。其中,RuII@Co3Fe-LDH(110)和PtIII@Co3Fe-LDH(110)催化剂过电位低至0.20 V。电子结构分析结果表明,TM掺杂M3N-LDH(110)体系可提高其导电性能,TM在不同位点的掺杂具有不同的电子调控效应。该文的第一作者为计算化学所2023级硕士研究生李世龙。此工作获得了国家自然科学基金面上基金项目的支持。