铂(Pt)与二氧化铈(CeO2)结合催化体系在逆水煤气反应(RWGS)中表现出优异的催化活性与选择性(Appl. Catal. B Environ., 2021, 291: 120101)。近期,计算化学研究所雷鸣课题组采用第一性原理理论计算研究了Pt掺杂二氧化铈(110)表面单原子催化剂(Pt1/CeO2-x(110) SAC)催化CO2还原为CO的RWGS反应机理(见图1),相关成果以全文发表在ChemCatChem上(ChemCatChem, 2025, 17(8): e202500027, https://doi.org/10.1002/cctc.202500027)。
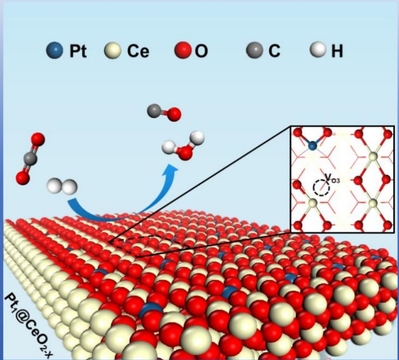
图1. Pt1/CeO2-x(110)单原子催化剂催化RWGS反应示意图。
本文研究中,氢气(H2)作为该反应的氢来源,我们首先探究了Pt1/CeO2-x(110)单原子催化剂催化RWGS反应的可能路径,包括CO2直接氧化还原机制和催化加氢机制,催化加氢机制又分为甲酸(HCOO*)机制和羧基(COOH*)机制(*表示为吸附状态)。
该文分别研究了Pt掺杂CeO2(110)的两种稳定结构(见图2),Pt1/CeO2(110)I和Pt1/CeO2(110)II。Pt1/CeO2(110)I基本保持了原始CeO2(110)的结构,而在Pt1/CeO2(110)II中,掺杂的CeO2(110)表面结构存在较大畸变,其中掺杂的Pt原子、两个表面O原子和两个次层O原子形成平面四配位结构。优化计算得到Pt1与CeO2(110)II的结合能(Eb)比Pt与CeO2(110)I的Eb更负,这表明Pt1/CeO2(110)II比Pt1/CeO2(110)I更稳定。
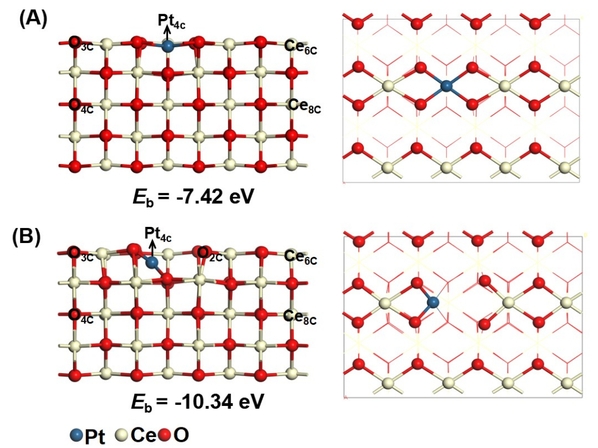
图2. (A)Pt1/CeO2(110)I与(B)Pt1/CeO2(110)II表面的侧视图和俯视图。
该文进一步研究了Pt1/CeO2(111)II表面的H2解离和氧空位的形成机制,这包含了A, B, C, D, E, F六个位点的反应路径(见图3)。计算结果表明,H2更倾向沿Path B在B位点发生异裂经TSB1过渡态生成HPt*和HO*(H4),然后通过氢转移机制经TSW3过渡态,生成水分子脱离后形成氧空位(VO)。
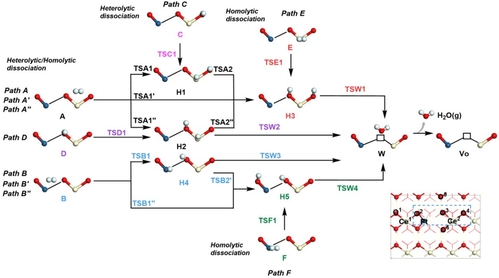
图3. Pt1/CeO2(110)II表面在A、B、C、D、E和F位点H2解离和氧空位形成的反应网络图。
Pt1/CeO2(110)表面随氧空位形成,形成了两个受阻Lewis酸碱对(FLP)活性位点:Ce2-O5(A位点)和Pt-O5(B位点)。该文研究了H2在含氧空位的Pt1/CeO2-x(110)表面的吸附和解离(见图4),计算结果表明,H2在Ce2-O5位点(A位点)发生均裂经TSA过渡态生成2HO*为优势反应路径。
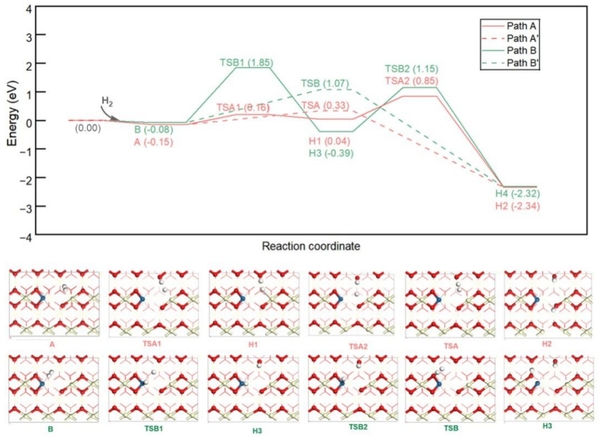
图4. H2在Pt1/CeO2-x(110)II表面发生解离反应的势能面图(单位:eV)及沿反应路径各驻点的俯视结构图。
随后,该文研究了二氧化碳(CO2)分子分别在Pt1/CeO2-x(111)II表面两个FLP位点(Pt-O5和Ce2-O5)和三个表面氧空位附近氧位点(O2、O5和O6)的吸附结构。基于反应物种在表面的结构变化、吸附能计算和电子结构分析,A、B和C构型表现出CO2的物理吸附行为,而C和D则表明CO2在催化剂表面的吸附为化学吸附。A、B和C构型中CO2呈线性吸附(li-CO2*),在B位点(Ce2-O5位点)吸附能最负。在D和E构型中CO2呈弯曲吸附(bt-CO2*),在D位点表现出CO2在Pt1/CeO2-x(111)II表面吸附的最稳定构型。
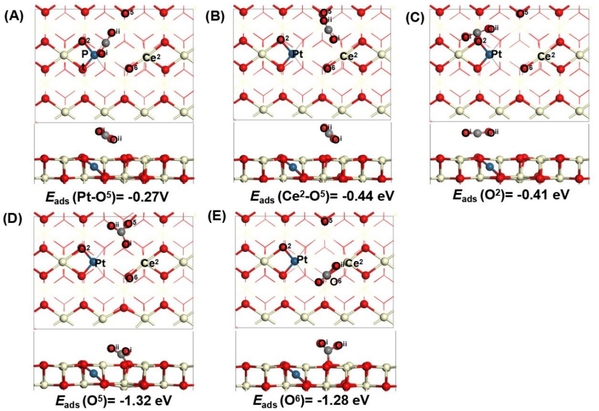
图5. Pt1/CeO2-x(110)II表面(A)Pt-O5、(B)Ce2-O5位点、(C)O2位点、(D)O5位点和(E)和O6位点处CO2吸附结构的侧视图和顶视图。
在Pt1/CeO2-x(110)表面催化RWGS反应机理研究中,如图6和图7所示,该文从两种CO2吸附构型(li-CO2*和bt-CO2*)开始,研究了RWGS反应的可能反应路径:(1) 传统的氧化还原机制(Path A和Path A’),(2)甲酸HCOO*机制(Path B和Path B’),和(3)羧基COOH*机制(Path C和Path C’)。计算结果表明,CO2加氢机制(COOH*和HCOO*机制)比传统的氧化还原机制更有利,Pt1/CeO2-x(110)II催化RWGS反应的最优势路径为从li-CO2*开始的羧基的Path C反应路径(li-CO2*→COOH*→CO*+OH*→CO*+H2O*),速率决定步骤为羧基COOH*(COOH*→CO*+OH*)的解离步骤(Ea为0.65 eV)。最后,本文采用从头算分子动力学模拟(AIMD)研究了在300K和700K下Pt1/CeO2-x(110)II催化剂的动力学行为,确认其结构的热力学稳定性。
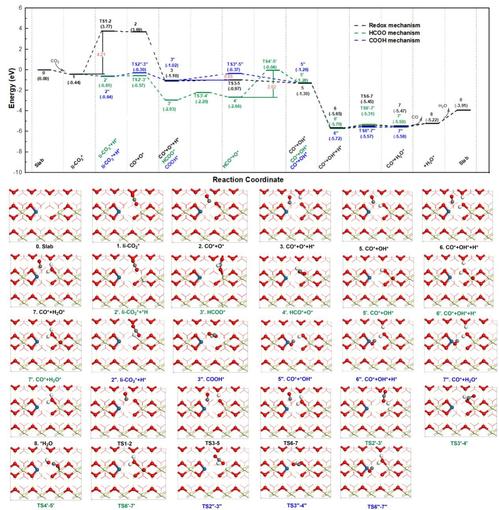
图6. Pt1/CeO2-x(110)II表面从li-CO2*构型开始的RWGS的Path A,B和C反应路径势能面图(单位:eV)及沿反应路径各驻点的俯视结构图。
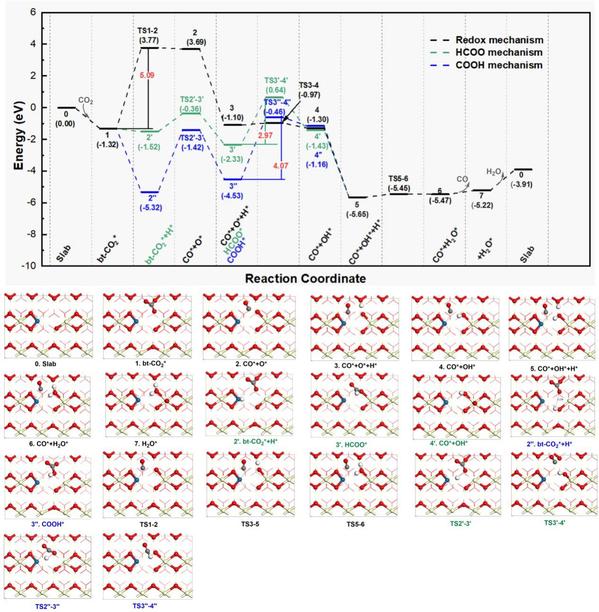
图7. Pt1/CeO2-x(110)II表面从bt-CO2*构型构型开始的RWGS的Path A’,B’和C’反应路径势能面图(单位:eV)及沿反应路径各驻点的俯视结构图。
综上所述,该研究采用第一性原理计算研究了Pt1/CeO2-x(110)单原子催化剂催化RWGS反应的机理,提出催化加氢路径比直接氧化还原路径更为优势,RWGS反应的最优势路径为线性吸附CO2构型(li-CO2*)开始的羧基反应路径(li-CO2*→COOH*→CO*+OH*→CO*+H2O*)。该理论研究工作为二氧化铈基催化剂表面催化RWGS反应的反应机理提供了重要的理论参考。该文的第一作者为计算化学所2022级硕士研究生盖莹同学。此工作获得了国家自然科学基金面上基金项目的支持。