氮气的活化与固定一直是实验和理论研究的重要领域之一。上个世纪初发展的Haber-Bosch过程将氮转化为氨(NH3),其中工业合成NH3产量的80%用于尿素的生产,是固氮工业生产中的重大突破。该工艺支持了全球一半以上的粮食生产,但该过程需要高温高压的苛刻反应条件,工业固氮每年消耗全球2%的能源并排放全球3%的CO2,其高能耗、低效率和环境污染等缺点也很明显,阻碍了经济的可持续发展。因此,发展反应条件温和、高效、环境友好的固氮技术具有非常重要的意义。其中,在温和条件下采用电催化方法催化氮还原反应(NRR)生产NH3过程是一类具有潜在应用前景的绿色催化过程。近期,北京化工大学计算化学研究所雷鸣课题组采用密度泛函理论(DFT)方法设计研究了15种过渡金属掺杂的120个双原子石墨氮化碳氮还原电催化剂(DAC,MM’@g-C6N6,M/M’=Ti,V,Cr,Mn,Fe,Co,Ni,Cu,Zn,Zr,Nb,Mo,Hf,Ta,W)催化NRR反应活性(见图1),采用3 + 1策略筛选出8个具有潜在优异活性和选择性的NRR电催化剂,为NRR电催化剂的理性设计提供了重要的理论参考。相关成果以全文发表在Chem. Mater.上 (https://doi.org/10.1021/acs.chemmater.5c00207)。
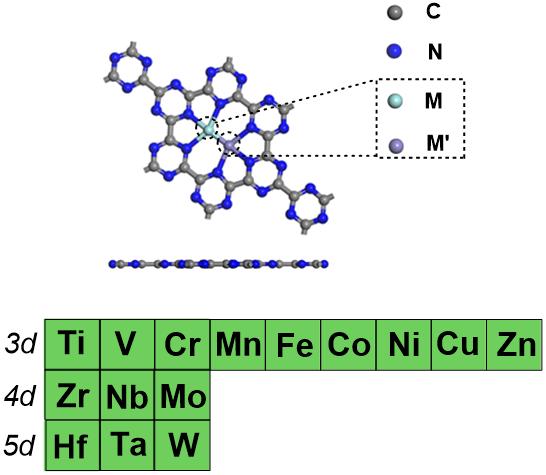
图1. 用于电催化氮还原反应(NRR)的双原子掺杂石墨氮化碳氮还原电催化剂(MM’@g-C6N6)结构示意图。
为快速高效筛选设计的MM’@g-C6N6的DAC电催化剂,本研究采用了3+1的高通量筛选策略(见图2)。1. 评估DAC的热力学稳定性,即金属原子掺杂在g-C6N6上的结合能(Eb)应小于0.0 eV (Eb < 0.0 eV);2. 评估N2吸附在MM’@g-C6N6表面上的吸附稳定性,即N2的吸附能(Eads(N2))应小于−0.50 eV (Eads(N2) < −0.50 eV);3. 考察DAC电催化NRR反应的潜在决速步骤(N2第一步氢化步骤(*N2+H→*N2H)和最后一步氢化步骤(*NH2+H→*NH3))的催化性能,即ΔG*N2→*N2H和ΔG*NH2→*NH3均小于0.30 eV;4. 对筛选出的DAC电催化剂进行全路径分析,并考察其NRR/析氢反应(HER)选择性,即比较NRR反应的限制电位(UL)与HER的UL值(UL(NRR)−UL(HER) > 0),确保其NRR选择性。
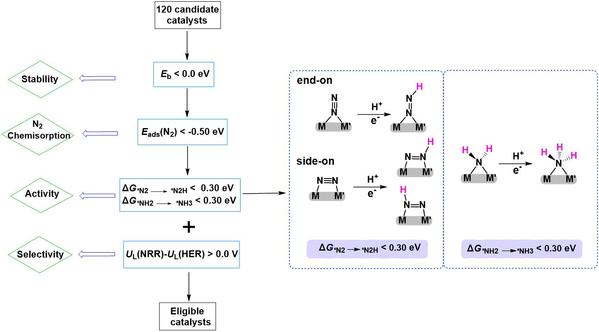
图2. 用于NRR的MM’@g-C6N6电催化剂的3+1筛选策略示意图。
催化剂结构的稳定性对催化剂活性有重大影响。Eb值用于研究M/M’原子与基底的结合强度,用于评估设计的MM’@g-C6N6电催化剂的热力学稳定性。本文首先研究了MM’@g-C6N6 (M/M’=Ti,V,Cr,Mn,Fe,Co,Ni,Cu,Zn,Zr,Nb,Mo,Hf,Ta,W)的Eb值,如图3热图所示,设计的MM’@g-C6N6电催化剂都表现出负的Eb值,优化得到的MM’@g-C6N6保持了相对稳定的二维平面结构,表明它们具有稳定的双原子掺杂结构。
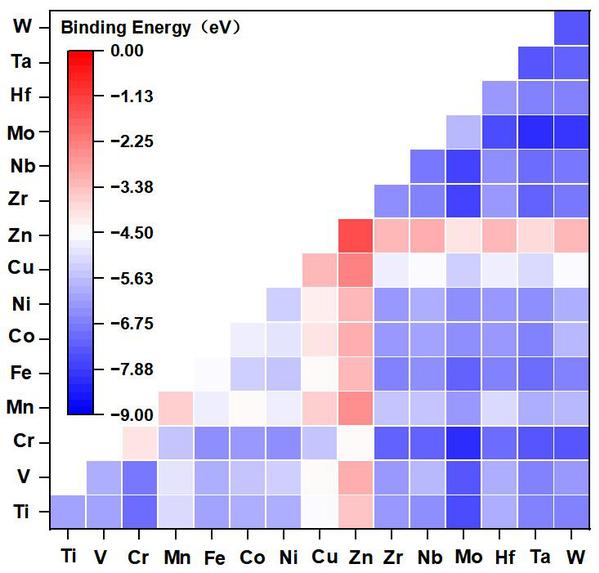
图3. 过渡金属(M/M’)原子掺杂后形成的MM’@g-C6N6电催化剂的结合能(Eb)热图(单位:eV)。
N2分子中N≡N惰性三键的活化需要较高能量,使得N2分子在催化剂表面上的吸附成为NRR过程中的关键步骤之一。本研究将N2在MM’@g-C6N6表面上的吸附能(Eads(N2)设定为小于−0.50 eV,考察N2在催化剂表面上的吸附强度。如图4a所示,N2可以以side-on吸附和end-on吸附模式吸附在构建的MM’@g-C6N6表面上。本研究分别计算了N2在MM’@g-C6N6表面上的side-on吸附模式和end-on吸附模式的吸附能(见图4b−4d)。根据Eads(N2) < −0.50 eV的筛选标准,筛选出88个side-on吸附模式和84个end-on吸附模式结构。
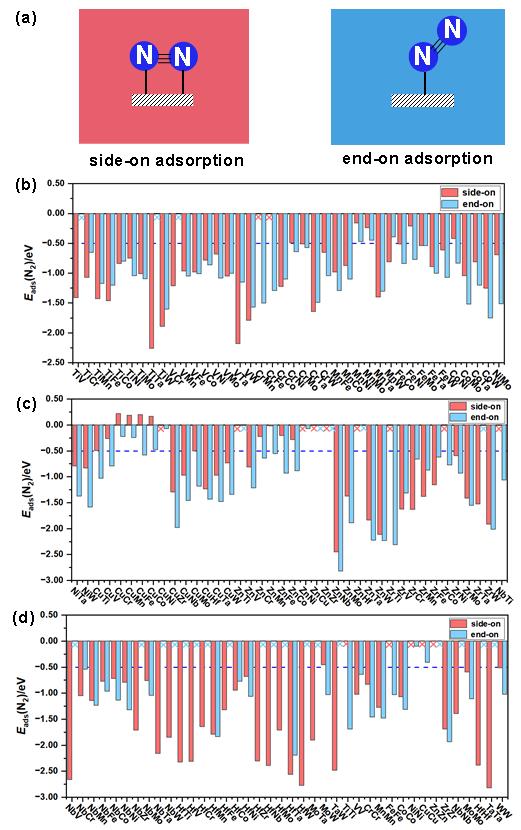
图4. (a) N2在MM’@g-C6N6表面的side-on吸附和end-on吸附模式。计算的N2在(b) TiV@g-C6N6到NiMo@g-C6N6、(c) NiTa@g-C6N6到NbTi@g-C6N6及(d) NbV@g-C6N6到WW@g-C6N6表面的吸附能(Eads(N2),单位:eV) (×表示该模式不存在)。
众多研究表明,对于大多数用于电催化NRR反应,第一步氢化步骤和最后一步氢化步骤(*N2+H++e-= *N2H,*NH2+H++e-= *NH3)为潜在的关键决速步骤,以ΔG*N2→*N2H和ΔG*NH2→*NH3均小于0.30 eV为筛选标准,可以显著提高筛选效率而不损失筛选精度。因此,本研究使用ΔG*N2→*N2H和ΔG*NH2→*NH3为活性描述符,筛选出13个采用side-on吸附模式和1个采用end-on吸附模式的MM’@g-C6N6催化体系(见图5)。
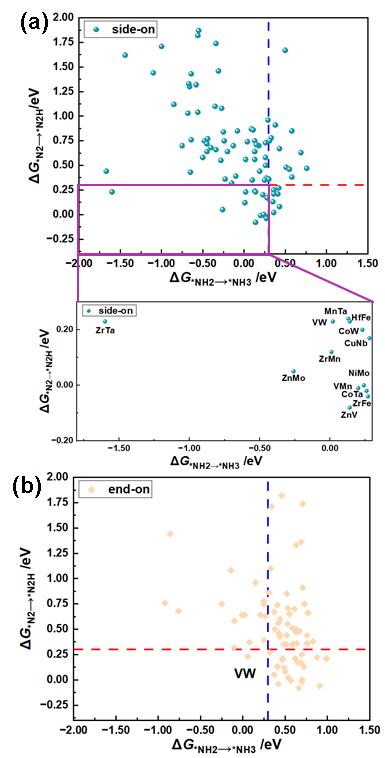
图5. 采用(a) side-on吸附模式(左下方区域为放大视图)及(b) end-on吸附模式的ΔG*N2→*N2H和ΔG*NH2→*NH3自由能变化值的比较图,用于研究MM’@g-C6N6催化NRR活性(单位:eV)。
如图6所示,NRR反应的分子反应机制包括了远端、交替、连续、酶促和混合机制。对于具有side-on吸附模式的DAC电催化剂,本文研究了其连续和酶促机制;而对于具有end-on吸附模式的DAC电催化剂,本文研究了其交替和远端机制。同时,混合机制将酶促/交替机制与连续/远端机制连接起来,在吸附模式下,*NH2NH和*NHNH2物种可以被氢化为*NH3和*NH物种,*NH可以进一步氢化形成最终的NH3产物。为进一步评价筛选出的MM’@g-C6N6电催化NRR性能,本文进一步系统研究了13个筛选出的MM’@g-C6N6电催化NRR全路径吉布斯自由能图。图7给出了活性最好的ZnV@g-C6N6和ZnMo@g-C6N6电催化的NRR反应吉布斯自由能图,最优势路径的UL值分别为−0.14 V和−0.15 V,指出连续/远端或混合路径更为优势。
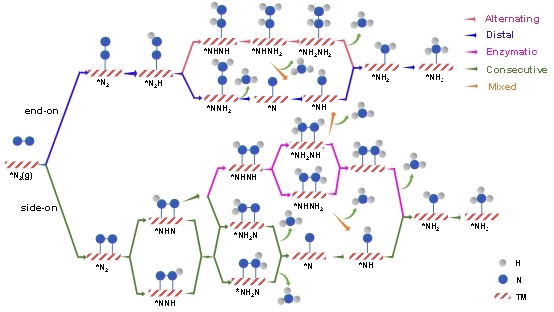
图6. MM’@g-C6N6电催化NRR的反应机理示意图。
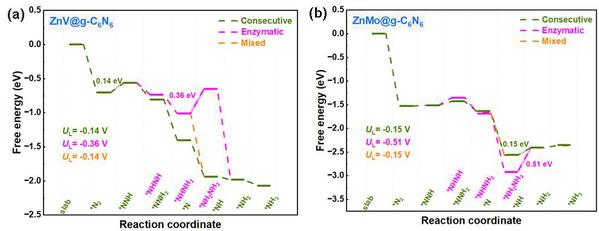
图7. 采用side-on吸附模式的(a) ZnV@g-C6N6和(b) ZnMo@g-C6N6催化NRR的吉布斯自由能图。
同时,在电催化NRR反应中,HER是一个重要的竞争反应,它会消耗氢质子并降低NRR的法拉第效率。因此,本文采用了NRR与HER之间的UL差异(UL(NRR)−UL(HER)>0)为标准,评估MM’@g-C6N6电催化NRR反应的选择性(见图8)。研究表明,八个DAC催化体系(ZnV@g-C6N6、ZnMo@g-C6N6、CoW@g-C6N6、VW@g-C6N6、NiMo@g-C6N6、CuNb@g-C6N6、MnTa@g-C6N6和HfFe@g-C6N6)均位于右下区域,表明NRR选择性更优。
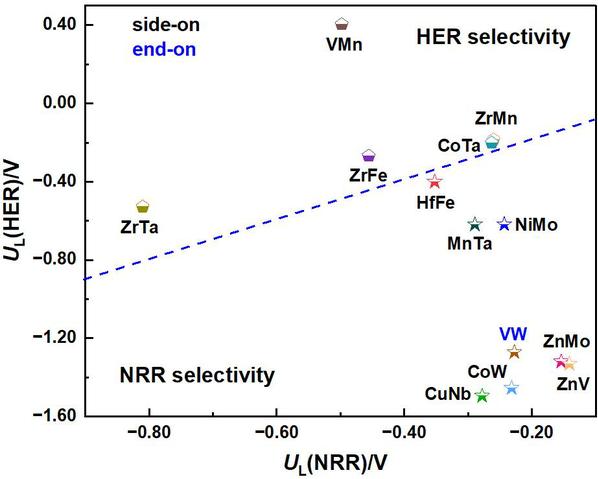
图8. 13个筛选出的MM’@g-C6N6的NRR与HER之间的UL值比较图(单位:V)。
最后,本研究构建了基于13个筛选的DACs催化的NRR的UL和ΔG*N的火山图关系(见图9),筛选出的ZnV@g-C6N6、ZnMo@g-C6N6、CoW@g-C6N6、VW@g-C6N6、NiMo@g-C6N6、CuNb@g-C6N6、MnTa@g-C6N6和HfFe@g-C6N6位于火山顶附近,ΔG*N范围从−1.5到0 eV,表明它们具有优异的NRR电催化活性。值得注意的是,ZnV@g-C6N6电催化剂最接近火山顶,具有适中的*N吸附吉布斯自由能(ΔG*N =−1.40 eV),表现出优异的NRR电催化活性。计算结果表明,ΔG*N值可以作为评估MM’@g-C6N6的NRR电催化活性的描述符。
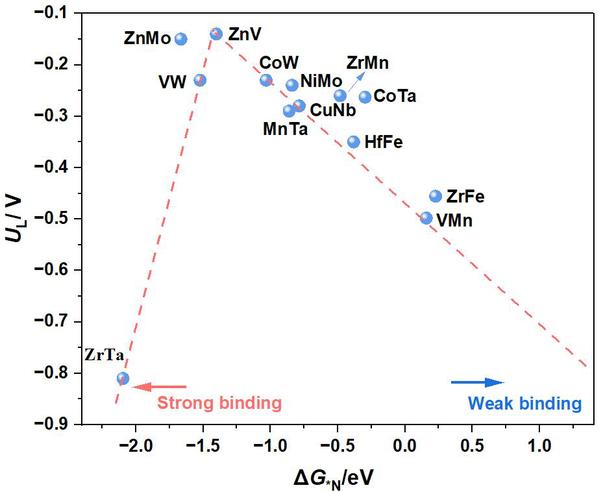
图9. 构建的筛选出的MM’@g-C6N6电催化NRR反应限制电位(UL)与*N吸附吉布斯自由能(ΔG*N)之间的火山图(单位:eV),向左的箭头表示*N的强吸附,向右的箭头表示*N的弱吸附。
综上所述,本研究设计了120个双原子掺杂石墨氮化碳NRR电催化剂MM’@g-C6N6,采用3+1筛选策略,系统地研究了MM’@g-C6N6电催化NRR反应活性,筛选出了8个具有优异电催化活性和选择性的DAC体系,其中ZnV@g-C6N6和ZnMo@g-C6N6电催化NRR的UL值仅为−0.14 V和−0.15 V,构建了ΔG*N与UL之间的火山图关系。本研究工作为NRR双原子电催化剂的理性设计与筛选提供了重要见解。该文的第一作者为计算化学所2023级硕士研究生崔洪云。此工作获得了国家自然科学基金面上基金项目的支持。